 What is too small to see with the naked eye, manufactured by half the population in batches of millions and with an alarming supply? The answer, according to some scientists, is sperm. Specifically, researchers are concerned that men in the West have been producing less and less sperm since the 1970s, a decline they say shows no signs of stopping. At the current rate, they say, these men could be infertile by 2045. But these numbers should make us think. The idea that men's sperm in Western countries is about to collapse is, in a word, extraordinary. The data does not support it.
The fear of a decrease in sperm count is powerful and powerful. It has been voiced by everyone from environmentalists like Erin Brockovich to white supremacists and their mainstream media spokespersons like Tucker Carlson. The great appeal of this notion is possible because the supposed causes of a decline span the entire material and ideological gamut. They include chemicals found in common household products, as well as modern urban lifestyles in which white men are physically sedentary and forced to share power with people of constantly diverse genders and ethnicities. It's worth taking a closer look at the evidence before heading to the "save the sperm" rally. The most recent round of apocalyptic predictions was sparked by an influential 2017 article that compiled sperm count data from studies published between 1973 and 2011. Figures for the US, Canada, Western Europe, Australia, and New Zealand, is that is, rich countries, mostly white, were grouped in the "western" category and the remaining data were grouped in the "other" category. While there was insufficient data to draw conclusions about "other" men, the authors found that the sperm count of the average population among men in the "Western" category had decreased by more than 50% since 1973. We analyzed the data and found that the apocalyptic verdict of the disappearance of the sperm is far from the only plausible interpretation of what is happening here. The authors assume that the high sperm counts of men in "Western" nations in the 1970s represent the norm. This assumption makes the pernicious but all-too-common mistake of treating the men of prosperous, white-majority nations as the standard by which everyone else should be compared. It also takes for granted that when it comes to sperm, the more the merrier. The available evidence does not support this association: male fertility does not scale proportionally with sperm count. Some men with a low sperm count can be very fertile, while others who are overflowing with sperm can have a hard time conceiving. Add to this the well-known fact that sperm count is very context sensitive (to tight underwear, exercise, a hot bath, and even the season) and it's easy to see how a single sperm count measurement is an unreliable indicator of fertility. . There is also the question of what is causing the decrease in sperm count. If we take seriously the idea that environmental pollutants are hostile to sperm production, we would expect to see the most drastic declines among men living in the most polluted environments. It is well established that the world's poor, that is, those who live predominantly (but not exclusively) in the "other" countries, bear the greatest burden of environmental pollution. However, the authors and the media have launched into framing the crisis as one that confronts "Western" men; what is ignored is the fact that the study data were insufficient to draw conclusions for men in the "other" category. The very use of the categories "western" and "other" makes little scientific sense and matters dangerous racial nuances. Men frequently migrate between "western" and "other" nations, making countries a poor indicator of environments that could affect any man's sperm. And conditions vary widely within nations, especially large and heterogeneous ones like the United States or Brazil. Knowing which passport a man is carrying tells you little about contaminants or other possible sperm-reducing factors he may have encountered. Apart from all this, these seemingly alarming findings may simply reflect normal variation. This would be unprecedented - studies have documented natural ups and downs in the levels of reproductive hormones such as testosterone and progesterone, with no impact on fertility. Could sperm counts vary in the same way? The researchers don't even consider this possibility. The lesson from sperm depletion research is not that we are facing imminent human extinction (at least not for sperm-related reasons). Rather, it is the most banal but accurate fact that there is much we do not know about the relationship between men's reproductive health and environmental pollution. This blind spot is what we should pay attention to. A long and sexist history of scientists zealously focusing on female reproduction has led researchers to neglect male fertility. The legacy of chemical industry lobbying and industry-funded research distorts our understanding of the effects of plastics exposure on human health. And a racist history of treating rich white male bodies as the norm of the species sets us up to ignore the majority of the world's population. In the face of all this uncertainty and obfuscation, what we need is better science: scientific institutions free from corporate influence and diverse researchers trained to unearth hidden racist and sexist assumptions. Our failure to meet these standards is the real reason for the panic. Source:https://www.theguardian.com/commentisfree/2021/jun/07/scare-stories-falling-sperm-counts-male-infertility-science
0 Comentarios
Daniel Beck 1, Millissia Ben Maamar 1, Michael K Skinner 2 BACKGROUND Over the past two decades, numerous studies have demonstrated a form of non-genetic inheritance called epigenetic transgenerational inheritance that is mediated by germline alterations in epigenetic processes [1-3]. One of the first observations involved vinclozolin, an environmental agricultural toxicant, which is one of the most widely used agricultural fungicides, to induce epigenetic transgenerational inheritance of testicular pathology and alterations in DNA methylation [1]. Similar observations with a wide variety of environmental toxicants, from dioxins to DDT (dichloro-diphenyl-trichloroethane), have identified similar epigenetic inheritance impacts in a variety of different diseases [3-5]. Transgenerational phenotypic manifestations of vinclozolin and DDT include induction of testicular, prostate, renal, and ovarian pathology, as well as obesity [3]. An early observation in mice identified a stress-induced traumatic impact on the epigenetic transgenerational inheritance of behavioral abnormalities [6, 7]. Interestingly, injection of eggs with the ncRNA from stressed individual male sperm promoted the same transgenerational phenotypes [6]. Subsequent studies have supported the role of cRNA or DNA methylation in germline-mediated epigenetic transgenerational inheritance [3, 8]. This epigenetic transgenerational inheritance phenomenon has been shown to be induced by environmental chemicals, nutrition, stress, and traumatic abnormalities in rodents and humans [3, 7, 9], as well as by a wide variety of environmental stresses in plants [10, 11], insects [12, 13], worms [14], fish [15-17], birds [18, 19] and a variety of mammals such as pigs and humans [20-22]. Various physiological impacts have been observed including pathologies in the brain, reproductive organs, kidneys, immunity, obesity, and infertility [1-3]. The phenomenon of environment-induced epigenetic transgenerational inheritance has been well established and has a significant impact on the etiology of disease [2, 3] and other areas of biology such as evolution [23]. Although most previous research has focused on a single epigenetic process such as DNA methylation [3, 4, 10] or ncRNA [6, 8], few have examined multiple processes. Our previous studies demonstrated in the inheritance of vinclozolin and DDT-induced epigenetic transgenerational pathology that the sperm of the transgenerational F3 generation had coordinately altered differential DNA methylation regions (DMR), noncoding RNA expression (ncRNA), retention sites of Differential histones (DHR) and histone modifications [24, 25]. These observations suggest potential interactions between the different epigenetic processes, but this remains to be elucidated during the phenomenon of epigenetic inheritance. Previous studies have shown a role for ncRNA in RNA-directed DNA methylation in several different systems [26-28]. NcRNA can help locate the DNA methylation site and facilitate subsequent chromatin remodeling processes. Therefore, the integration of ncRNA and DNA methylation has been established. Histone modifications can also be dramatically modified by remodeling of cRNA and chromatin from euchromatin-active gene expression sites to heterochromatin-inactive DNA sites [29]. Although information is available on histone retention in sperm and its impact on the embryo [30, 31], the potential role of different epigenetic processes in histone retention has not been reported. Recently, a role for environmental exposures (eg, Vinclozolin and DDT) has been observed in promoting transgenerational epigenetic inheritance of sperm histone retention [24, 25, 32]. The current study investigates the potential integration of DNA methylation, ncRNA, and histone alterations in the phenomenon of epigenetic transgenerational inheritance. Previous analyzes of the concurrent expression of epigenetic processes between F1, F2 and F3 generations with a strict statistical threshold have shown negligible overlap between different generations or between epigenetic processes [24, 25]. The current study used an extended overlap analysis with a less stringent statistical threshold and found overlaps between generations and epigenetic marks. The potential integration of the different epigenetic processes and generational conservation was identified. RESULTS The experimental design involved pregnant female Sprague Dawley rats of the F0 generation at 120 days of age exposed during embryonic days 8-14 (E8-E14) transiently to vinclozolin (100 mg / kg of body weight / day) or DDT (25 mg / kg body weight / day), or vehicle dimethyl sulfoxide (DMSO) control, as previously described [24, 25]. The offspring of the F1 generation were obtained and aged to 90 days of age and then raised within the lineage (control, vinclozolin or DDT) to generate the large offspring of the F2 generation. Subsequently, the F2 generation was bred in a similar manner to generate the large F3 transgenerational offspring within the lineage. In each generation or lineage the rearing of siblings or cousins was not used to avoid any artifact of consanguinity [1, 3]. Litter bias was avoided by euthanizing litters at 10 (approximately 5 females and 5 males), and then only one or two males and females from each litter were used for reproduction within the lineage, as described above. All males were up to 120 days old and were sacrificed for sperm collection for molecular analysis, as described in previous studies [24, 25]. The number of individual animals investigated in each generation for sperm collection and molecular analysis was approximately 10 to 17 males, so n = 10 to 17 for animals with three different groups of 4 to 6 animals for each generation and analysis. epimutation. The collected sperm were used to isolate RNA, DNA, and chromatin for ncRNA analysis, DNA methylation, histone retention, and histone modification, as described in previous studies [24, 25], (Fig. 1). Molecular data from these previous studies (GEO # GSE109775, GSE106125 and NCIB SRA: PRJNA430483 largeRNA (control and DTT), PRJNA430740 smallRNA) were analyzed to explore the data more bioinformatically. 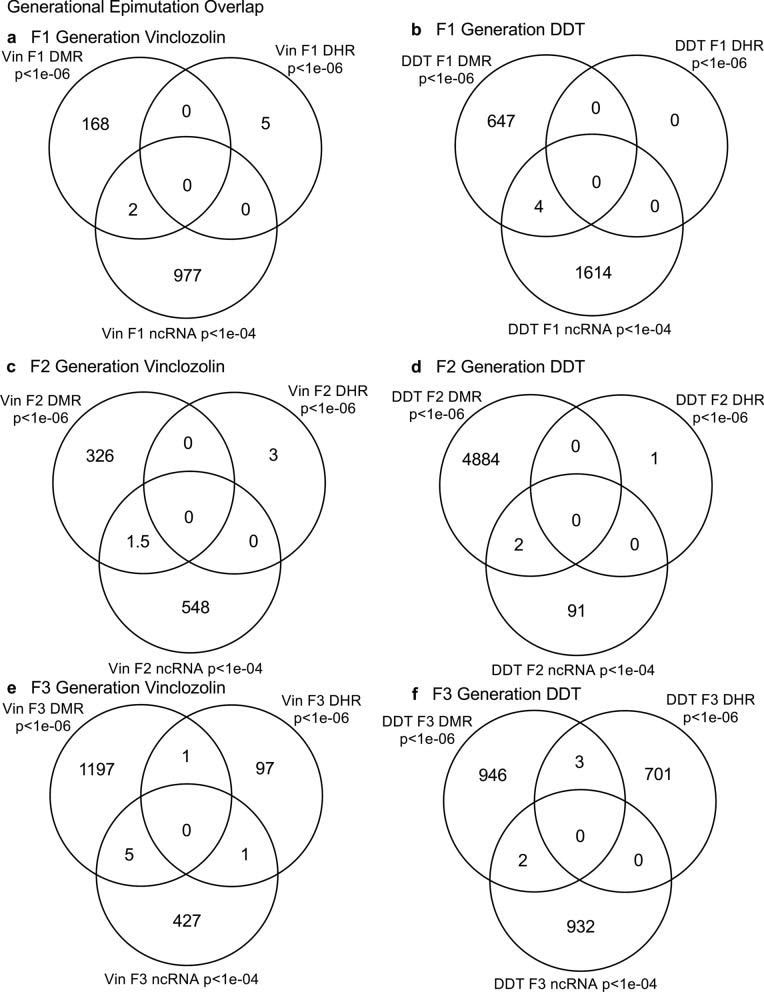 Fig. 1 Generational epimutation overlap at high stringent statistical threshold. a F1 generation vinclozolin lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). b F1 generation DDT lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). c F2 generation vinclozolin lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). d F2 generation DDT lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). e F3 generation vinclozolin lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04). f F3 generation DDT lineage DMR (p < 1e−06), DHR (p < 1e−06), and ncRNA (p < 1e−04) Sperm DMR, ncRNA (both small sncRNA and large lncRNA) and DHR were analyzed in each sample, as previously described [24, 25], for the vinclozolin and DDT generation sperm samples F1, F2 and F3. The numbers and overlaps of DMR, ncRNA and DHR for each generation with a high stringency threshold are presented as previously reported in (Fig. 1). The overlap with a Venn diagram for the transgenerational F3 generation for the different epigenetic marks is negligible at the high stringency threshold, (Fig. 1e, f), for each exposure, as previously identified [24, 25]. The F1 and F2 generations were also mainly distinct between the epimutations (Fig. 1a-d). Although different epigenetic alterations are present in each generation for both exposure lineages, the overlaps with a strict statistical threshold were insignificant, suggesting different functions and a lack of integration, as previously suggested [24, 25]. Interestingly, when a highly stringent epimutation was compared with the others at p <0.05, several genomic locations were identified with the different types of epimutations present.The chromosomal locations of these altered epigenetic marks (ie, epimutations) are presented in (Fig. 2) and in (Additional File 1: Tables S1-S6) for each generation for vinclozolin and DDT lineage sperm samples. Color-coded labels identify DMR, ncRNA, and DHR in all genomes with common chromosomal locations for each generation. Only those significant sites in high stringency (color code index) are shown with an analysis of epimutations that overlap with the other epimutations at p <0.05, (Fig. 2). Specific epimutation chromosomal locations, statistical p-values and gene associations are presented in (Additional File 1: Tables S1-S6). In the F1 and F2 generations, only alterations were found in the ncRNAs and DMRs, as previously described [24, 25]. Therefore, overlaps mainly occurred between ncRNA and DMR in the F1 and F2 generations (Fig. 2a-d). DHRs were developed in the transgenerational F3 generation, as previously described [24, 25]. In the F1 and F2 generations, the ncRNA was predominantly high statistically significant epimutation and overlap with DMR at p <0.05, (Fig. 2a-c), with a mixture of ncRNA and DMR in the F2 generation of the DDT lineage, (Fig. 2d). The transgenerational F3 generation also had a mixture of ncRNA and DMR with high statistical significance, as well as a series of DHRs (Fig. 2e, f). Therefore, chromosomal locations with multiple epimutations are identified with ncRNA predominant in the F1 and F2 generations with the high statistical threshold, and DMR being more predominant in the F3 generation with a mixture of the various epimutations (Fig. 2 and file Additional 1: Tables S1-S6). 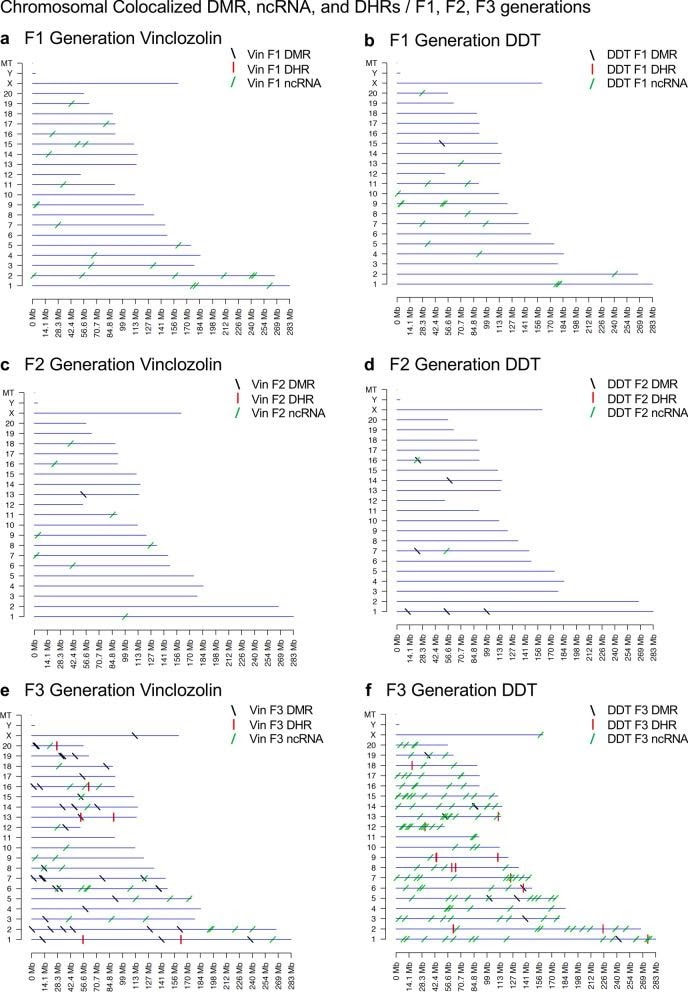 Fig. 2 Chromosomal colocalization of overlap epimutations. The overlap of one epimutation at high statistical stringency (DMR p < 1e−06, DHR p < 1e−06, or ncRNA p < 1e−04) overlap with others at p < 0.05. The epimutation at high stringency is identified with color and marked as indicated by the inset legend. The chromosomal number and size (megabase) are presented. a F1 generation vinclozolin lineage ncRNA and DMR. b F1 generation DDT lineage ncRNA and DMR. c F2 generation vinclozolin lineage ncRNA and DMR. d F2 generation DDT lineage ncRNA and DMR. e F3 generation vinclozolin lineage DMR, DHR and ncRNA. f F3 generation DDT lineage DMR, DHR and ncRNA An extended overlap analysis was performed with the DDT and vinclozolin lineage data using a less stringent statistical threshold for comparisons (Fig. 3). The strictest statistical threshold epigenetic data sets (DMR p <1e-06, ncRNA p <1e-04, and DHR p <1e-06) were compared between generations and epigenetic marks with a statistical threshold p <0.05 . This optimized the potential to identify overlaps compared to the more stringent thresholds used in (Fig. 1). The rows present the strictest DMR, ncRNA, and DHR thresholds for the F1, F2, and F3 generations. The columns present the corresponding threshold overlaps p <0.05 with the highest p-value threshold data sets. Examining the horizontal rows, as expected, shows 100% overlap (that is, shaded) for the same data set and the number of associated epigenetic marks and the percentage (%) of overlap with the left margin value. .This extended overlap allows the two different stringency thresholds to be compared and additional observations of overlap to be determined (Fig. 3). Similar trends are seen in the overlaps for the DDT and vinclozolin data sets. One of the initial observations was that the ncRNA from the F1 generation had a high percentage of overlap with the DMR from the F3 generation (Fig. 3 and Additional File 1: Tables S1 and S2). Similar observations are made with the F1 and F2 generations. For the F1 generation, the DDT ncRNA had more than 20% overlap observed with the F1, F2 and F3 generation DMRs, while the F1 vinclozolin ncRNA had approximately 35% overlap with the F1 generation DMRs. F2 and F3 (Figs. Figs. 33 and and 4a, 4a, b). Lists of overlapping ncRNA and DMR sites are presented in (Additional File 1: Tables S1 and S2). The F2 and F3 generation ncRNAs were similar with the overlap with the DDT generation DMRs of approximately 20%, but it was reduced to 10-15% with the vinclozolin DMRs, (Fig. 3). Therefore, some ncRNAs were common between generations and had an overlap with DMRs varying between 8 and 35% overlap for vinclozolin DMRs and 15-20% overlap for DDT DMRs. The possibility that ncRNA may promote RNA-directed DNA methylation is suggested. The Venn diagrams presented in (Fig. 4a, b) support these overlays and the epigenetic overlays of ncRNA and DMR are listed in (Additional File 1: Tables S1 and S2). 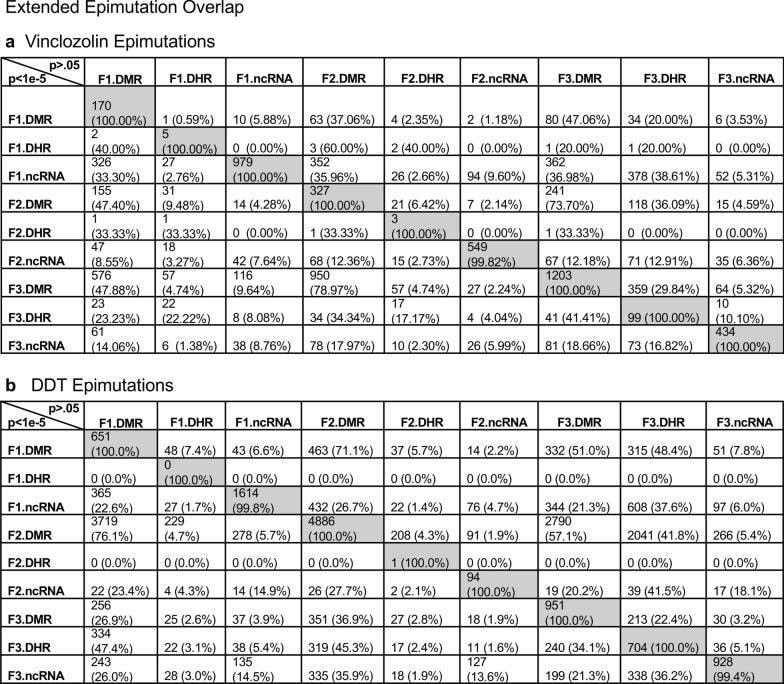 Fig. 3 Extended epimutation overlap. The epimutations at high stringency (DMR p < 1e−06, DHR p < 1e−06, and ncRNA p < 1e−04) in rows were compared to epimutations at p < 0.05 in columns. The number of overlap epimutations and percentage of the total are presented for each overlap. As anticipated, 100% overlap was observed for the same generation and epimutation indicated by shaded box. a Vinclozolin lineage epimutation and b DDT lineage epimutation overlap 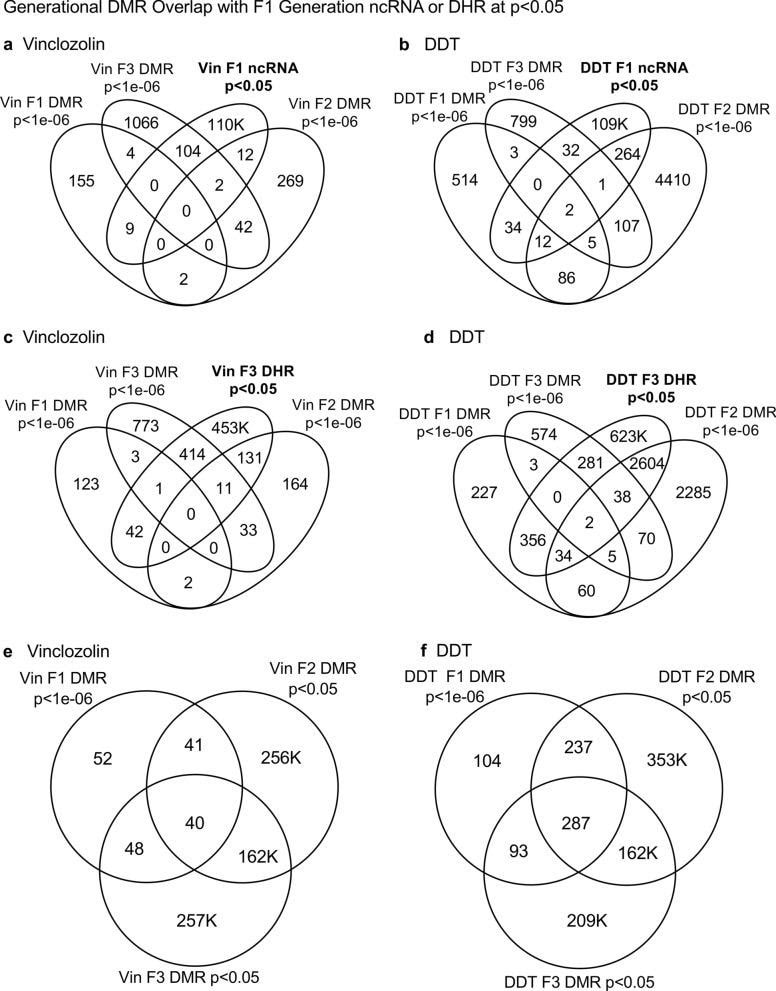 Fig. 4 Epimutation overlaps. Generational DMR overlap with F1 generation ncRNA p < 0.05. A Venn diagram overlap of F1, F2, and F3 generation DMR (p < 1e−06) with F1 generation ncRNA (p < 0.05). a Vinclozolin lineage DMR and ncRNA overlap. b DDT lineage DMR and ncRNA overlap. Generational DMR overlap with F3 generation DHR p < 0.05. A Venn diagram overlap of F1, F2, and F3 generation DMR (p < 1e−06) with F3 generation DHR (p < 0.05). c Vinclozolin lineage DMR and DHR overlap. d DDT lineage DMR and DHR overlap. Generational DMR overlap. A Venn diagram overlap of F1 generation DMR (p < 1e−06) with F2 and F3 generation DMR (p < 0.05). e Vinclozolin lineage DMR overlap. f DDT lineage DMR overlap The next observation was that the DMRs of the F1, F2 and F3 generation had a 20-48% overlap with the DHRs of the F3 generation for the DDT and vinclozolin lineages (Fig. 3). Interestingly, the DHRs of the F3 generation had a 23-47% overlap with the DMRs of the F1, F2 and F3 generations for both challenge lineages. The Venn diagram overlaps in (Fig. 4c, d) supports these overlays of DMR and DHR and suggests that DMRs may help guide DHR formation transgenerationally. The overlapping DMRs and DHRs of the F3 generation are presented in (Additional file 1: Tables S3 and S4). An interesting observation was the overlap between the F1, F2 and F3 generation DMRs for DDT and vinclozolin exposures (Figs. (Figs. 33 and 4e, 4e, f). The highest overlap for the F1 generation DMRs of DDT was the F2 DMR from the F3 generation with 71% overlap, and for the vinclozolin F2 DMRs with the F3 generation DMRs with 73% overlap. The highest for the F3 generation DMRs was a 79% overlap with the vinclozolin F2 DMRs. Generally, 25-50% There was overlap between the F1, F2 and F3 generation DMRs for both exposures, (Fig. 3). A Venn diagram supports this observation and demonstrates approximately 25% overlap for vinclozolin DMRs and 35% overlap for DDT DMRs, (Fig. 4e, f) Lists of these overlapping DMRs are presented in (Additional File 1: Tables S5 and S6). Therefore, a percentage (25-35%) of the individual DMRs of the F1 generation were retained transgenerationally. Mind. Generally, the epigenetic alterations of the F3 generation had more overlap with each other and with the other generations for both exposures. Venn diagram analysis was used to identify epigenetic sites with overlapping DMR, ncRNA and DHR (Fig. 4). The overlapping F3 generation epigenetic sites were approximately 25% for the vinclozolin and DDT lineages. A permutation analysis was performed to show that this is significantly greater than the observed random overlap, with a p-value of p ≤ 0.05 for the 1 kb and 10 kb overlap sites. Several sites were randomly selected and mapped to identify the overlapping chromosomal locations of DMR, ncRNA, and DHR (Fig. 5). The actual statistical significance of the overlapping epimutations in these examples includes: (Fig. 5a) (ncRNA p <1e-04, DHR p <0.03 and DMR p <0.005); (Fig. 5b) (ncRNA p <1e-04, DHR p <0.001 and DMR p <0.0004); (Fig. 5c) (DHR p <1e-08, ncRNA p <0.005 and DMR p <0.04); and (Fig. 5d) (ncRNA p <1e-06, DMR p <1e-04 and DHR p <1e-05). The "Discussion" section reviews the possibility of RNA-directed DNA methylation and DMR-directed histone retention.  Fig. 5 Genomic colocalization of DMR, DHR and ncRNA. The genomic and colocalized DMR, DHR and ncRNA presented. The region size (bp), genes present, and localization of DMR, DHR and ncRNA identified. The various examples include a nc-005100.4, b nc-005104.4, c nc-005111.4, and d nc-005113.4 from the NCBI Rattus norvegicus release 106 in 2016 All previous analyzes and overlays presented were based on a direct overlapping chromosomal location for ncRNA, DMR, and DHR. The question of whether there are a greater number of sites with epimutations that are in the same region but do not directly overlap was addressed. A distance of 5 kb was used on each side of the epimutations to have a 10 kb window for the region of potential overlap. An extended overlap was used using this 10 kb window with the same data that will identify the directly overlapping and nearby sites within the 10 kb window, Fig. 6. The level of overlap with a 10 kb window identified the Same overlays presented and discussed, but the level of overlap was in the range of 80 to 99% (Fig. 6). Both the vinclozolin and DDT lineages had the same high level of overlap, with the majority being a range> 90%, with a permutation analysis p value of p <0.001. Using this 10 kb window, most of the ncRNA, DMR, and DHR overlapped between generations and epimutations. This supported all previous observations and demonstrated a significant level of epimutation overlap. Since approximately 90% of the F3 generation DMRs overlapped with the F3 generation DHR and the F1 generation ncRNA, the conserved F3 generation DMRs (Additional File 1: Table S5 and S6) were used in a Pathway Studio analysis to link genes associated with DMR processes and cellular pathologies, (Additional file 1: S1 and S2). A large number of genes associated with DMR and epimutation linked to various previously observed transgenerational pathologies, including kidney disease, breast tumors, immune abnormalities, prostate disease, metabolic disease, or behavioral abnormalities [3,33,34]. 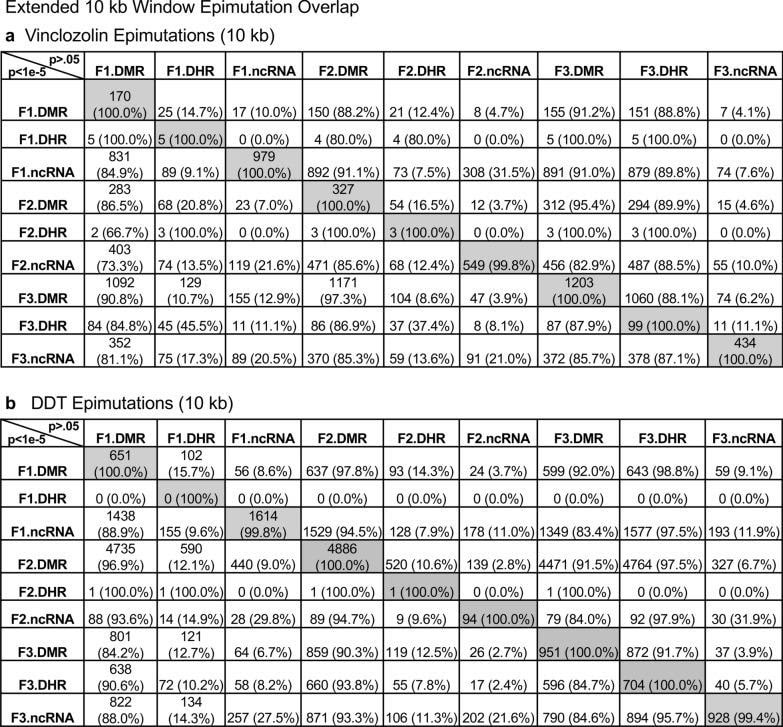 Fig. 6 Extended epimutation overlap within a 10-kb region. The epimutations at high stringency (DMR p < 1e−06, DHR p < 1e−06, and ncRNA p < 1e−04) in rows were compared to epimutations at p < 0.05 in columns. The number of overlap epimutations and percentage of the total are presented for each overlap. As anticipated, 100% overlap was observed for the same generation and epimutation indicated by shaded box. a Vinclozolin lineage epimutation and b DDT lineage epimutation overlap . DEBATE Previous studies have demonstrated the concurrent presence of DMR, ncRNA, and DHR in sperm after exposure to DDT or vinclozolin in pregnant females of the F0 generation during gonadal sex determination [24, 25]. These data were obtained and reported on a strict statistical threshold selection and demonstrated negligible overlap in each generation (Fig. 1) [24, 25]. The current study was designed to further investigate the potential integration of different epigenetic processes between the F1, F2, and F3 generations. An approach was adopted to compare the strictest statistical threshold values for DMR, ncRNA, and DHR with the least stringent threshold p <0.05 between different epigenetic processes and generations. This extended overlap approach generated a series of observations to suggest intergenerational integration for the epigenetic transgenerational inheritance phenomenon. An interesting observation of extended DMR overlap demonstrated that approximately 40-50% of F1 generation sperm DMRs were retained and were also present in transgenerational F2 and F3 generations (Figs. (Figs. 33 and 4e, 4e, f). This was 88-97% of the DMRs when 10 kb windows were used, (Fig. 6). A permutation analysis showed this to be significant (p <0.001) and not due to random associations. Conserved DMRs of the F1 generation in subsequent generations is presented in (Additional file 1: Tables S5 and S6, and those DMRs with gene associations suggest that approximately 50% of these conserved DMRs were associated with genes. Many of these genes had associations with a variety of pathologies, Additional file 1: Figures S1 and S2) Therefore, a percentage of the F1 generation sperm DMRs were programmed and then preserved in subsequent generations. s DMR of sperm from the F1 generation were generationally conserved, there were minimal similarities between the different generations of ncRNAs, (Figs. (Figures 33 and 6) 6). DHRs were present mainly in the sperm of the F3 generation, so they were not conserved between generations (Fig. 3). In contrast, when a 10 kb region is considered approximately 40% of the DHRs of the F3 generation are present in the F1 and F2 generations, (Figure 6). The potential role of these DMRs for guided transgenerational histone retention is discussed below. The second interesting observation was the overlap of F1 generation sperm ncRNA with F1, F2 and F3 generation DMRs. More than 20% in DDT and 35% in vinclozolin F1 generation ncRNA overlap with DMR generation F1, F2 and F3, (Figs. (Figs. 33 and and and 44 and additional file 1: Tables S1 and S2). Observations suggest the potential role of RNA-directed DNA methylation in direct exposure F1 generation and transgenerational F3 generation Previous literature has established a role for RNA-directed DNA methylation in various biological and cellular systems [26-28] This implies the ability of cRNA to recruit or target chromatin remodeling proteins and proteins such as DNA methyltransferase to guide DNA methylation at a chromosomal site, which has been established in a variety of different organisms and developmental processes [26 -28]. Observations suggest that ncRNA-directed DNA methylation may play a role in the phenomenon of epigenetic transgenerational inheritance. Although the ncRNA of the F1 generation has the highest supe In comparison with the DMRs of the F3 generation, there are also overlaps with the F1 and d cRNAs of the F2 generation with the DMRs of several generations, (Fig. 3). When considering an overlap of the 10 kb region, the ncRNAs of the F1 generation have a 91% overlap with the DMRs of the F2 and F3 generations, (Fig. 6). The overlays of ncRNA and DMR suggest that ncRNA-directed DNA methylation has a potential role in the epigenetic transgenerational inheritance process (Fig. 7). A combination of F1 generation direct exposure alterations in ncRNA and subsequent transgenerational F3 generation actions on DNA methylation appears to be involved. Epigenetic sites colocalized with ncRNA and DNA methylation support this proposal (Fig. 5). Although the molecular process of RNA-directed DNA methylation has been established [26-28], and has been suggested in generational impacts in plants and humans [35, 36], the current study only demonstrates the strong correlations of ncRNA and the DMR. Future studies are needed to provide further molecular insights and validation of ncRNA-directed DNA methylation in the transgenerational epigenetic phenomenon 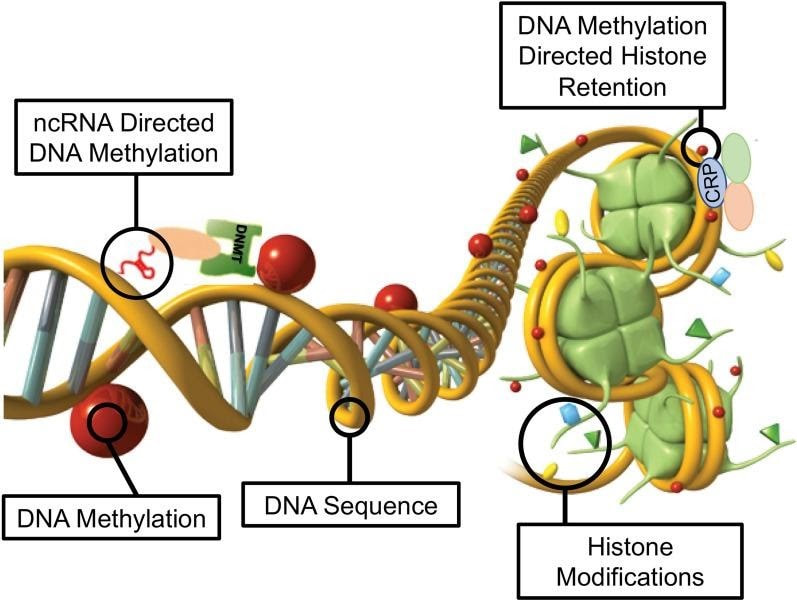 Fig. 7 Diagram of ncRNA-directed DNA methylation and DNA methylation-directed histone retention. The red dot identifies DNA methylation, green histone the nucleosome with modifications in histone tails indicated. The ncRNA association with cofactors and DNA methyltransferase (DNMT) promoting DNA methylation (red dot) for RNA-directed DNA methylation. The DNA methylation (red dot) association with chromatin remodeling proteins (CRP) to promote histone retention is indicated Another interesting observation was the overlap of the DMRs of the transgenerational F3 generation with the DHRs. Although negligible DHRs are present in the F1 or F2 generations, the F3 generation has DHRs that overlap with the DMRs of the F1 and F2 generations (Fig. 3). For DDT DMRs there was a 35-50% overlap and for vinclozolin DMRs, a 23-41% overlap. Considering an overlap of regions of 10 kb, the DHRs of the F3 generation had an 85-95% overlap with the DMRs in all generations (Fig. 6). Permutation analysis demonstrated that this number of 10 kb region overlaps is not due to random associations (p <0.001). The literature on the spermatid exchange of histones for protamines to condense DNA in the head of spermatozoa is well established in most of the organisms investigated [37-39]. Although the vast majority of sperm DNA has associated protamines, a percentage of histones is retained, ranging from 5% to 10% of DNA in different mammalian species [40]. Previously, we found that histone retention was significantly increased in sperm of the transgenerational F3 generation with the presence of new retention sites [24, 25, 32]. Therefore, an additional epigenetic mechanism influenced during the epigenetic transgenerational inheritance process involves altered histone retention [32]. The previous literature has described transition proteins and the processes of substitution of histones by protamines [41, 42], but the role of epigenetic processes such as DNA methylation has not been considered. Our previous observations suggest a role for this process in epigenetic inheritance [24, 25]. The current study indicates a potential role for DNA methylation in guiding or directing histone retention (Figs. Retention, (Figs. 33 and 6) .6). Previous studies have shown a critical role for DNA methylation in the actions of chromatin remodeling proteins [41-43]. So DNA methylation could alter associated proteins and secondary structure of DNA which is one aspect of the histone retention process. Although more investigation of molecular processes is required in future studies, observations from the current study suggest a potential role for differential histone retention driven by DNA methylation (Fig. 7). It is proposed that DMRs help guide or direct histone retention sites so that a greater number of sites appear transgenerationally. Therefore, the existence of histone retention directed by DNA methylation is proposed, and the observations support a transgenerational integration of DMR and DHR. An additional interesting observation is that the F1 and F2 generation DMRs that develop after direct exposure to toxic substances are similar to the F3 generation DMRs, but that the DHRs were not formed until the transgenerational F3 generation (Figures 33 and 66).
The results of the current study help to integrate previous data obtained with ncRNA, DMR and DHR [24, 25]. The potential roles of ncRNA-driven DMRs and DMR-driven DHRs are suggested. A percentage of the DMRs from the F1 generation are retained and conserved for subsequent F2 and F3 generations. The F1 generation ncRNA overlapped with the F2 and F3 generation DMRs, supporting the role of ncRNA-directed DNA methylation and DMR formation (Fig. 7). The specific subtypes of sncRNA and lncRNA in this process will require further investigation. The potential of DMR-driven DHRs is suggested, but more information is required to elucidate the specific processes involved. About half of the overlapping epimutations had known associated genes. Many of these genes are associated with previously identified pathologies (Additional file 1: Figures S1 and S2), thus supporting a transgenerational pathology mechanism. The proposed model and the integration of the transgenerational ncRNA, DMR and DHR are presented in (Fig. 7). The observations of the current study suggest the integration of epigenetic processes in the transgenerational epigenetic phenomenon. It provides insights into the development and generational transmission of these environmentally induced sperm epimutations that have previously been shown to be associated with the development and etiology of the disease. The potential use of these integrated epigenetic chromosomal sites as biomarkers to identify exposure and / or susceptibility to disease suggests that they could be used as diagnostics to facilitate preventive medicine in the future. More research is needed to further establish these mechanisms in the phenomenon of epigenetic transgenerational inheritance, but the current study provides support and a framework for the integration of various epigenetic processes. CONCLUSION Observations with the two different exposures of DDT or vinclozolin suggest that the generational impacts and transgenerational integration of ncRNA, DMR, and DHR are similar. A variation in the percentage of overlaps is observed, but the same trends and integration conclusions of the various epimutations are similar for the DDT and vinclozolin exposure lineages. The colocalized epimutation sites for the different exposures demonstrate the same phenomenon, but independent sites are observed for each exposure. The two different models of environment-induced epigenetic transgenerational inheritance support the proposed general mechanism for ncRNA-directed DNA methylation and DMR-directed development of DHR. Although the current study identifies such co-localized and interacting epimutation sites, many of the specific ncRNA, DMR, and DHR are not co-localized [24, 25]. Therefore, the independent actions of ncRNA, DMR, and DHR will also be important in the mechanism involved in environment-induced epigenetic transgenerational inheritance. A combination of ncRNA, DMR and DHR epimutations developed during gametogenesis allows post-fertilization embryonic impacts and suggests that integration of ncRNA and DMR will be involved in epigenetic inheritance. The mechanism proposed in (Fig. 7) helps to elucidate the molecular mechanisms involved in the epigenetic transgenerational inheritance phenomenon. SUMMARY OF MATERIALS AND METHODS Studies and animal husbandry As described previously [24, 25] and expanded upon in (Additional File 1: Supplemental Methods), exogenous Sprague Dawley SD male and female rats were fed a standard diet with water ad lib and mated. Pregnant female rats were exposed to DDT or vinclozolin, and the pups were reared within each lineage for three generations in the absence of exposure. The F3 generation was aged up to 120 days for sperm isolation and molecular analysis, as described in (Additional File 1: Supplemental Methods). Sperm were isolated and used for epigenetic analysis, as described in (Additional file 1: Supplementary methods). All experimental protocols for rat procedures were previously approved by the Washington State University Animal Care and Use Committee (IACUC protocol # 6252), and all methods were performed in accordance with relevant guidelines and regulations. Epigenetic, statistical and bioinformatics analysis As previously described [44], DNA was isolated from sperm collected at the time of dissection. The DNA isolation protocol has been described previously [33,34], (Additional file 1: Supplementary methods). Methylated DNA immunoprecipitation (MeDIP), followed by next generation sequencing (MeDIP-Seq) was performed on the isolated DNA. MeDIP-Seq, sequencing libraries, next-generation sequencing, and bioinformatic analysis were performed, as described previously [33, 34] and in (Additional File 1: Supplementary Methods). All molecular data has been deposited in the public database at NCBI (GEO # GSE109775 and GSE106125), and R-code computational tools are available on GitHub (https://github.com/skinnerlab/MeDIP-seq) and https: // skinner .wsu.edu / genomic-data-files-and-r-code /. Since mid-May 2020, the CDC has been tracking case reports of multi-system inflammatory syndrome in children (MIS-C), a rare but serious condition associated with COVID-19. CDC is working to learn more about why some children and teens develop MIS-C after having COVID-19 or coming into contact with someone with COVID-19, while others do not. As of October 1, 2020, the number of patients meeting the MIS-C case definition in the United States exceeded 1,000. In 2021, this number surpassed 2,000 on February 1 and 3,000 on April 1. Last update with cases reported to CDC on or before May 3, 2021 *: Additional patients are being investigated. After review of additional clinical data, patients may be excluded if there are alternative diagnoses that explain their disease. Since mid-May 2020, the CDC has been tracking case reports of multi-system inflammatory syndrome in children (MIS-C), a rare but serious condition associated with COVID-19. CDC is working to learn more about why some children and teens develop MIS-C after having COVID-19 or coming into contact with someone with COVID-19, while others do not. As of October 1, 2020, the number of patients meeting the MIS-C case definition in the United States exceeded 1,000. In 2021, this number surpassed 2,000 on February 1 and 3,000 on April 1. Last update with cases reported to CDC on or before May 3, 2021 *: Additional patients are being investigated. After review of additional clinical data, patients may be excluded if there are alternative diagnoses that explain their disease.  Characteristics of MIS-C patients being closely monitored by CDC by race and ethnicity, sex, and age. To date, the majority of MIS-C patients have been of Hispanic / Latino race / ethnicity or non-Hispanic black. Hispanic / Latino and non-Hispanic black populations have also been disproportionately affected by COVID-19 in general. Further studies of MIS-C are needed to learn why certain racial or ethnic groups may be disproportionately affected and to understand the risk factors for this disease. 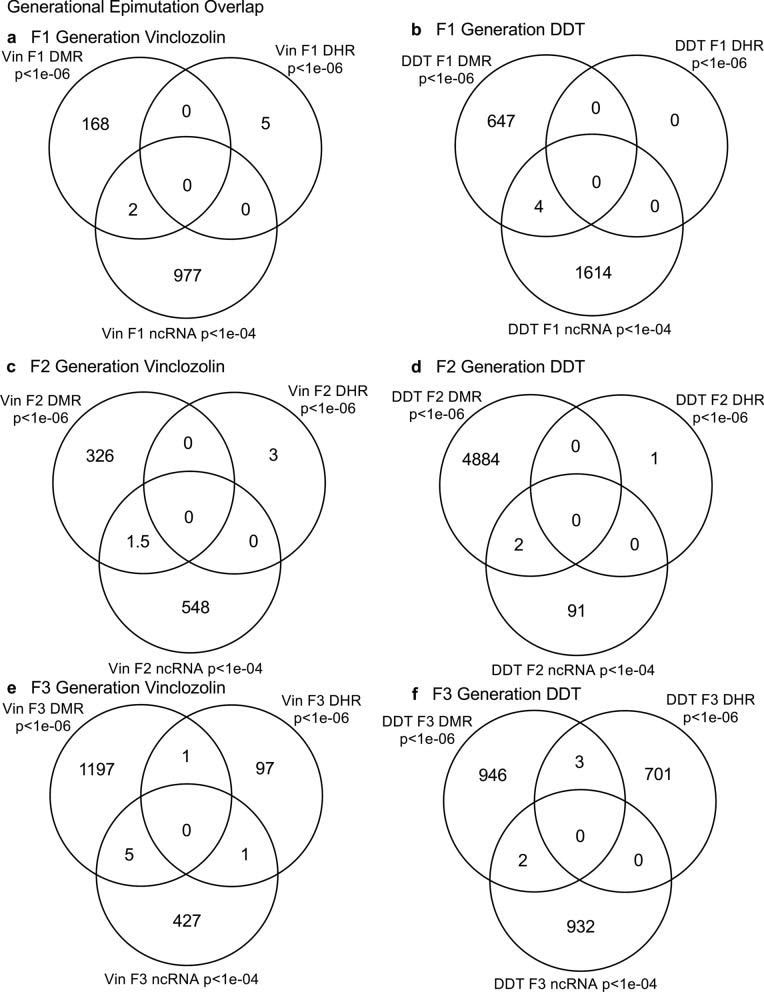 Next steps
MIS-C can occur weeks after COVID-19 and even if the child or family did not know the child had COVID-19. CDC and state authorities will be monitoring additional cases and will adapt the MIS-C recommendations as necessary. Researchers are evaluating reported cases of MIS-C and associated health outcomes to try to learn more about specific risk factors for MIS-C, disease progression in children and adolescents, and how to better identify MIS-C. and distinguish it from other similar ones. diseases. Additional comments Some patients may meet partial or full criteria for Kawasaki disease, but must be reported if they meet the MIS-C case definition Consider MIS-C in any pediatric death with evidence of SARS-CoV-2 Infection Timing of reporting Case reporting may be delayed due to limited capacity in state / local health departments and as the CDC evaluates the data to ensure that cases meet the MIS-C case definition. Source: https://www.cdc.gov/mis-c/cases/index.html |
fucobiWe are an organization at the service of environmental health working for the conservation and recovery of our natural resources in defense of human health. CategoriesArchives
Octubre 2022
|
- FUCOBI
- Español
- Awards
-
ONE HEALTH
- Student Projects
- mangroveENCODE >
-
ShrimpENCODE
>
- Shrimp Biodiversity and Genetic Diversity
- TSV in Shrimp
- WSSV in shrimp
- Shrimp allergens
- Metals / Cadmium in Shrimp
- Biodiversity and Genetic Diversity Fish
- Shell Genetic Diversity
- Crab Genetic Diversity
- Shrimp Epigenome Project
- Low level endocrine disturbing chemical potentials — changes in the expression of genes causing shrimp allergy in humans and other endocrine disturbing chemicals
-
childrenENCODE
>
- Contact
- Events
- publications
- How can you help
- Our Blog
- Portovelo
 Canal RSS
Canal RSS
